殷赋科技:让DS处理吧 。 用UCSF Chimera的Prep Dock就可以处理 , 我们平台也会自动处理蛋白 , 生成的mol2文件就含有质子化氢和偏电荷(partial charge) 。
F:你用什么做QMMM?
A:你好 , 我用的ds@F 。 下载的pdb晶体里面有一个配体很大 , 为了对接 , 清理蛋白质的时候定义活性位点设定的半径要不要相应增大啊?
文章图片

文章图片
G:不知道具体的结合位点?
A:我就选中的原来配体设定的半径 。
H:原来的配体就是活性位点 , 一般半径设置10左右吧 , 半径太大的话对接时间会长 , 另外结果不准确 , 当然半径太小会对接不进去 , 可以适当调整 。
A:你好 , 我看教程也是说半径设为10但是10的话我看无法全部包括原来的配体分子啊 。
H:你可以适当增大 , 12也可以 。
I:你们用的什么软件?我之前对接一个蛋白 , 用的原配体口袋 , 对接我的化合物打分挺高的 , 可是作用的氨基酸跟文献差好多 , 这正常么?
A:ds , 我主要是现在想要选一个好的晶体 , 把自带的配体对接回去和本身的配体重叠查看一下rmsd值 。
G:就是对接出来的结果不是那么准确吧 。
A:好多晶体对接回去都不好 , 我现在调大点半径试试 , 先对上再说 。
I:我之前用薛定谔对接的效果不错 。
A:我感觉你那种情况好像也挺正常 , 这种对接也不怎么准 , 尤其是ds 。
I:后来用的DS对接效果一般 , 但是作用的蛋白跟我用薛定谔对接的差不多 。
A:薛定谔好像是更靠谱一些 。
I:做起来挺慢的 。
A:你们有薛定谔的教程吗?
I:我是跟别人学了两天 , 也没太搞明白 。 原来学校有个课题组做这个方向的 , 我毕业走了 , 我们学校开始开的计算机辅助药物设计这门课了!
QSAR叠合构象 R:请问 , 做QSAR分子叠合的时候 , 有些叠合的不好该怎么搞呢 , 明明都差不多 。 殷赋科技:你指的是QSAR模型的预测能力不好吧?如果叠合不整齐 , 手动调整咯 。
R:你是说调训练测试集嘛?
殷赋科技:你是说结构叠合得不好(不整齐)吗?还是叠合后做出来的模型预测能力不好?
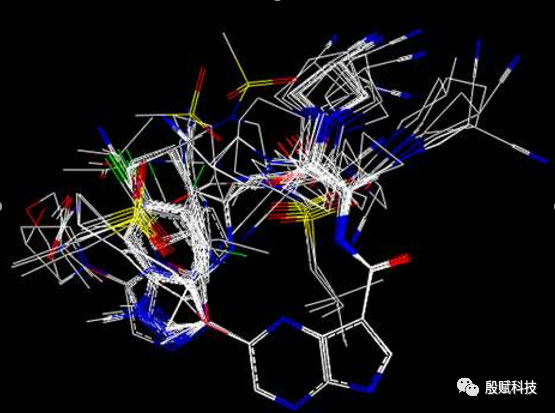
R:叠合出来的预测能力还行 confa的0.6几 , comsia的有0.7几 , 就是叠合的不整齐 。
殷赋科技:截图看如何不整齐 。
文章图片
殷赋科技:你是不是认为有部分结构应该左右反过来叠合才算整齐?
R:反倒是没反 。
殷赋科技:那就没问题啊 。
G:这是什么软件啊?
殷赋科技:SYBYL 。
R:有几个明明就一点差别 , 但是叠合出来就重合的不好 。
殷赋科技:那是构像问题了 , 它已经尽力了 , 但叠合是刚性的 , 构像如此 , 它也很无奈啊 。 调整构像呗 , 可以用你认为合适的某个化合物来构建相似结构的结构 , 构像就接近啦 。
殷赋科技:或者产生大量构像 , 然后用算法去挑选 。 我不知道SYBYL里边有没有这个功能 。
R:我刚看了教程找到了你说的利用构象搜索去挑选叠合构象@殷赋科技?感谢你的提醒 。
虚拟筛选 S:导师让我做虚拟筛选这块儿(学autodock), 但是没有头绪 , 我也不怎么会python , 请问有这方面的课程么?
殷赋科技:我们博客上有个脚本 , 可以用用 , 不过 , 并不建议新手去用脚本做筛选 。 因为计算不是问题 , 问题是没有经验的人的命中率很低 , 浪费的是买化合物做实验的钱 。
特别声明:本站内容均来自网友提供或互联网,仅供参考,请勿用于商业和其他非法用途。如果侵犯了您的权益请与我们联系,我们将在24小时内删除。